Edición genética CRISPR-Cas9: aplicaciones clínicas en 2025
By Carmen Vidal Salgado · 9 de mayo de 2026
Revisión del estado de CRISPR-Cas9 en ensayos clínicos 2025: entrega, seguridad, regulación y coste en anemia falciforme, beta-talassemia, distrofia muscular, cáncer y terapia de la vista.
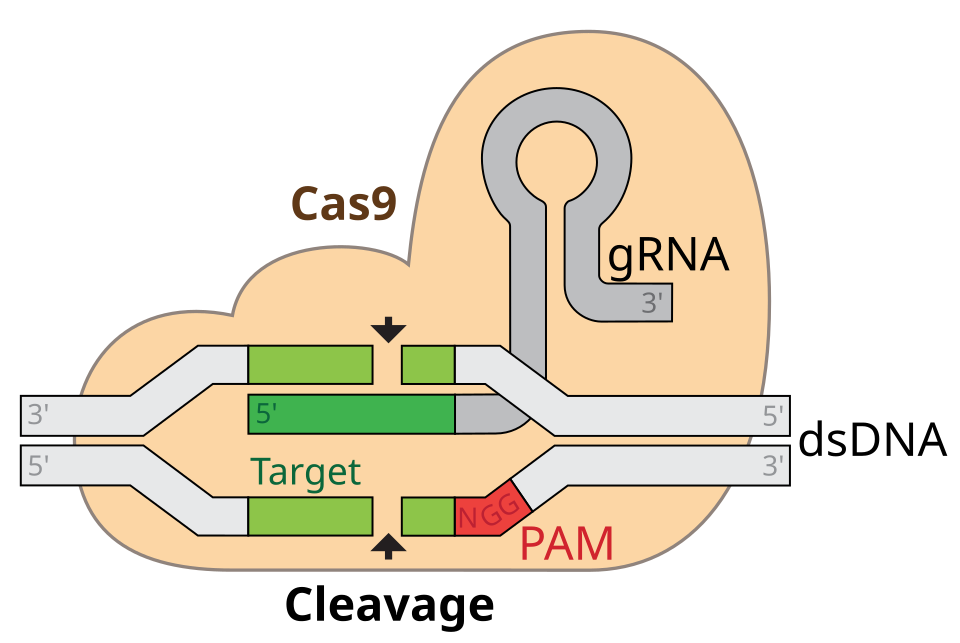
GRNA-Cas9 (Autor: marius walter · Licencia: CC BY-SA 4.0 · Fuente: Wikimedia Commons)
La edición genética CRISPR-Cas9 ha pasado de ser una hazaña tecnológica a una plataforma de intervención clínica en etapas tempranas de desarrollo, con un número creciente de ensayos en pacientes y un marco regulatorio en evolución. Este artículo sintetiza el estado de la edición CRISPR-Cas9 en 2025, poniendo foco en los ensayos clínicos para anemia falciforme y beta-talassemia, distrofias musculares, cáncer mediante CAR-T editado y terapias oculares, así como en vectores de entrega, seguridad y costes, y regulación. La evidencia clínica, fortalecida por los resultados de CTX001/Exa-cel que condujeron a la aprobación de Casgevy por la FDA en 2023, ha impulsado un crecimiento sostenido de las pruebas en múltiples indicaciones y contextos terapéuticos (Frangoul et al., 2021).
Panorama actual de los ensayos clínicos CRISPR-Cas9 en 2025
La investigación clínica con CRISPR-Cas9 ha pasado de demostrar la viabilidad de la edición a evaluar su eficacia, seguridad y viabilidad logística en pacientes. En anemia falciforme y beta-talassemia, la estrategia dominante es la edición ex vivo de células hematopoyéticas para activar la expresión de la hemoglobina fetal mediante la alteración de los reguladores genéticos; esta aproximación ha mostrado mejoras clínicas relevantes en cohortes tempranas y ha respaldado la aprobación regulatoria de Casgevy por la FDA en 2023 (Frangoul et al., 2021). En este marco, los ensayos continúan ampliando la selección de pacientes, ajustando protocolos de acondicionamiento y optimizando la persistencia de las células editadas.
En distrofia muscular y otras enfermedades monogénicas, la edición CRISPR-Cas9 se ha trasladado desde modelos preclínicos hacia ensayos en fases iniciales, con enfoques que incluyen la corrección de exones específicos o la interrupción de vías patológicas. Aunque la mayor parte de estos datos es previa a la validación clínica amplia en humanos, los avances en la optimización de la especificidad y la reducción de off-targets han aumentado la confianza en la viabilidad de estas estrategias para futuras indicaciones (Doudna & Charpentier, 2014).
En oncología, la edición de CAR-T cells para mejorar la persistencia, reducir la toxicidad y modificar rutas inmunometabólicas ha pasado de experimentos preclínicos a ensayos clínicos en etapas tempranas, con múltiples grupos evaluando combinaciones de edición, como la interrupción de PD-1 o la modificación de receptores de antígeno para ampliar el reconocimiento tumoral. Estos enfoques pretenden superar limitaciones de CAR-T convencionales y abrir nuevas candidaturas terapéuticas (Frangoul et al., 2021). Veremos que la complejidad de estos productos exige no solo eficacia sino control estricto de seguridad y manufactura, dada la naturaleza ex vivo de la edición y la necesidad de estandarización de procesos.
La terapia de la vista via CRISPR-Cas9 ha progresado hacia ensayos en retina, con intentos de corregir mutaciones causales de enfermedades hereditarias o de modular vías de degeneración. Aunque aún permanece en fases iniciales, este terreno destaca la posibilidad de intervenciones in vivo o ex vivo con diferentes plataformas de entrega, y complementa el abanico de órganos diana evaluados en CRISPR-Cas9. En conjunto, el estado de la edición CRISPR-Cas9 en 2025 refleja un aprendizaje progresivo sobre seguridad, eficacia y logística que condiciona la extensión de estas terapias a nuevos pacientes y a nuevas indicaciones (Doudna & Charpentier, 2014).
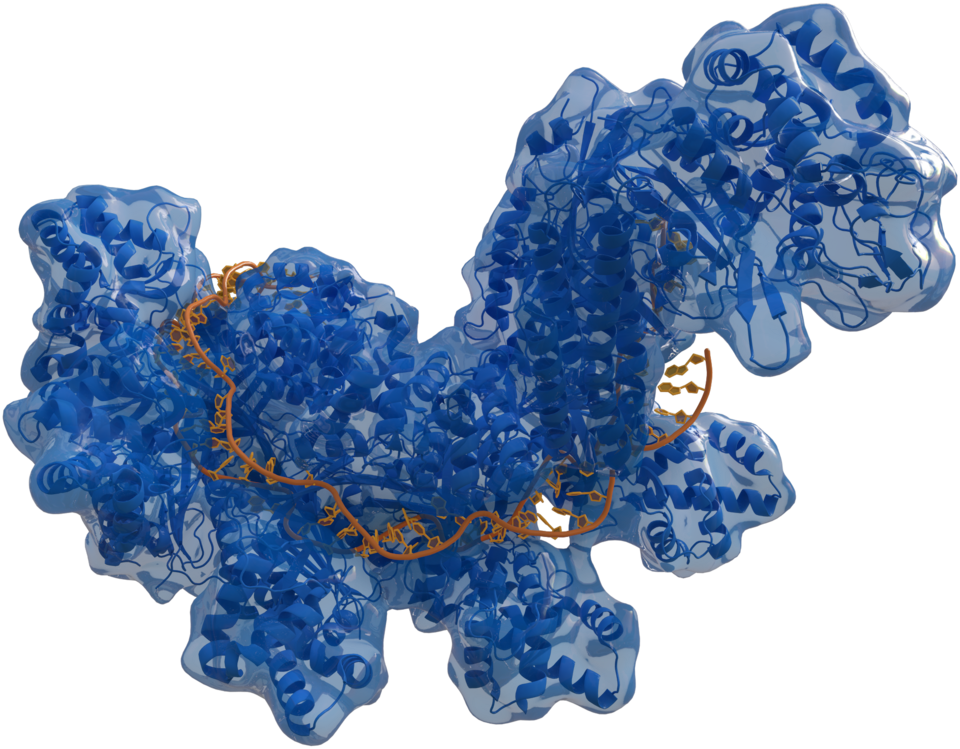
Vectores de entrega y enfoques de edición: AAV, LNP y abordajes ex vivo CAS 4qyz (Autor: Thomas Splettstoesser ( www.scistyle.com ) · Licencia: CC BY-SA 4.0 · Fuente: Wikimedia Commons)
La entrega de CRISPR-Cas9 es un cuello de botella técnico importante. En el ámbito ex vivo, las células hematopoyéticas pueden editarse fuera del cuerpo y luego reintroduirse, lo que facilita el control de la edición y la evaluación previa a la reinfusión. Este enfoque es particularmente relevante para hematología y enfermedades genéticas de la sangre, donde la ex vivo edición reduce la exposición del tejido sano a la nucleasa y facilita la selección de células adecuadas para el trasplante autólogo. En paralelo, los vectores in vivo –principalmente AAV y nanopartículas lipídicas (LNP)– buscan distribuir la edición en tejidos objetivo, pero presentan desafíos de especificidad, tamaño del cassette y respuesta inmunitaria.
Los vectores AAV han sido históricamente útiles para entregar componentes CRISPR en tejidos como retina y hígado; sin embargo, la limitación de capacidad y las respuestas inmunitarias han impulsado el desarrollo de variantes más eficientes y de estrategias de cassette reducidas. Las LNP han mostrado ventajas en entrega sistémica y han permitido aplicaciones en órganos como el hígado y, en algunos casos, la médula ósea cuando se combinan con enfoques de administración local. En 2024-2025, la investigación se ha centrado en mejorar la especificidad, reducir la carga inmunitaria y optimizar la cinética de expresión para minimizar off-targets y efectos adversos (Tsai et al., 2015; Kleinstiver et al., 2016).
La edición ex vivo de HSPCs (células madre hematopoyéticas) ha sido uno de los pocos enfoques con mayor grado de madurez clínica, sostenido por la posibilidad de evaluar la edición antes de la reinserción y de calibrar el proceso de trasplante. En otros tejidos, la entrega in vivo exige una solución a largo plazo para evitar respuestas inmunitarias repetidas y controlar la duración de la expresión de Cas9; en este sentido, el desarrollo de Cas9 de alta fidelidad y variantes de nucleasas con menor probabilidad de cortes no deseados ha sido crucial para la seguridad a largo plazo (Kleinstiver et al., 2016).
Seguridad, off-target y mejoras en la especificidad DNA animation (Autor: brian0918 ™ · Licencia: Public domain · Fuente: Wikimedia Commons)
La seguridad y la especificidad de CRISPR-Cas9 siguen siendo temas centrales para la aprobación clínica y la aceptación regulatoria. La detección y cuantificación de off-target edits, así como la evaluación de mutaciones indeseadas a nivel del genoma, son componentes clave de los ensayos clínicos. Métodos como GUIDE-seq, DISCOVER-seq y otras aproximaciones han permitido mapear posibles sitios de corte fuera del objetivo y guiar la optimización de guías y de variantes de Cas9 con mayor fidelidad (Tsai et al., 2015).
La generación de nucleasas de alta fidelidad y la ingeniería de guías con menor propensión a off-target han reducido el riesgo en escenarios clínicos. Ejemplos clásicos incluyen variantes de Cas9 con mutaciones que disminuyen cortes no deseados sin sacrificar la eficiencia en el objetivo, lo que se traduce en perfiles de seguridad más favorables para pacientes (Kleinstiver et al., 2016). Aun así, la evaluación de seguridad debe realizarse en cada indicación y en el contexto del tejido diana, la dosis y la duración de la expresión de Cas9, especialmente en aplicaciones in vivo.
En el caso de ensayos de hematología y terapia de la vista, la preocupación principal reside en la integridad del genoma de células editadas y en la posibilidad de mutaciones clonas que podrían originar efectos adversos tardíos. La combinación de enfoques de entrega más controlados y de Cas9 de alta fidelidad representa un camino razonable para mitigar estos riesgos sin comprometer la eficiencia terapéutica (Frangoul et al., 2021).
Regulación, costes y consideraciones éticas
La regulación de las terapias CRISPR-Cas9 varía entre la FDA en Estados Unidos y agencias equivalentes en la UE, con énfasis en la seguridad a corto plazo, la durabilidad de la respuesta clínica y la calidad de la fabricación. La aprobación de Casgevy (Exa-cel, CTX001) por la FDA en 2023 marcó un hito regulatorio, enfatizando la necesidad de controles de proceso riguroso y de evidencia de beneficio sostenido frente a riesgos. En Europa, EMA y agencias nacionales han seguido de cerca estos desarrollos y exigen evidencia de beneficio clínico estable y perfiles de seguridad compatibles con productos de alto coste y compleja logística de fabricación (Frangoul et al., 2021).
Los costes de estas terapias, que incluyen la edición de células autólogas, la manufactura para cada paciente y el monitoreo postratamiento, plantean desafíos para la adopción generalizada. Los modelos de pago basados en valor y la negociación de precios a largo plazo han sido propuestos como estrategias para equilibrar la inversión en innovación con la sostenibilidad de los sistemas de salud. Además, las consideraciones éticas en edición germinal o de tejidos germinales siguen siendo objeto de debate, aunque la mayoría de los ensayos clínicos se concentran en células somáticas para evitar impactos hereditarios no deseados (Doudna & Charpentier, 2014).
Perspectivas y retos para 2026 y más allá
El futuro de la edición CRISPR-Cas9 clínica depende de la consolidación de la seguridad a largo plazo, la mejora de la eficiencia en tejidos difíciles y la ampliación de indicaciones, preservando al mismo tiempo la viabilidad económica para sistemas de salud. La combinación de edición ex vivo en hematología, entrega in vivo en tejidos específicos mediante vectors optimizados, y estrategias de control de expresión ofrece un marco integrador para ensayos en hematología, ocular y oncología. La experiencia acumulada en 2023-2025, junto con avances en fidelidad de Cas9 y evaluación de off-target, augura una trayectoria de progreso gradual más que de rupturas súbitas, con avances clínicos que podrían ampliar el acceso a terapias génicas personalizadas (Jinek et al., 2012; Kleinstiver et al., 2016; Tsai et al., 2015).
La edición de CRISPR-Cas9 representa una plataforma que, bien gestionada, puede convertir enfermedades genéticas en condiciones controlables, pero exige un marco regulatorio robusto y una manufactura escalable para su adopción generalizada. (Doudna & Charpentier, 2014).
En síntesis, 2025 marca un punto de inflexión en la transferencia de CRISPR-Cas9 desde la demostración en laboratorio hacia ensayos clínicos con impacto real en pacientes. El equilibrio entre eficacia clínica, seguridad a largo plazo, viabilidad económica y aceptación regulatoria definirá el ritmo de adopción de estas terapias en la próxima década. El aprendizaje continuo de la experiencia clínica, respaldado por una investigación fundamental sólida sobre especificidad y entrega, será crucial para convertir el potencial de CRISPR-Cas9 en un componente estable de la medicina personalizada.
La edición de genes mediante CRISPR-Cas9 emergió como una plataforma versátil para la modificación del genoma en múltiples sistemas biológicos, con potencial clínico significativo.
Referencias clave citadas en el texto: (Frangoul et al., 2021); (Doudna & Charpentier, 2014); (Jinek et al., 2012); (Kleinstiver et al., 2016); (Tsai et al., 2015).
Carmen Vidal Salgado
Analista de investigación científica especializada en biología celular y técnicas de imagen.
Carmen Vidal Salgado escribe sobre avances en biología celular, técnicas experimentales y bioinformática para una audiencia académica de habla hispana. Su trabajo se enfoca en sintetizar investigación reciente — desde microscopía de super-resolución hasta edición genética CRISPR — en formato accesible pero técnicamente riguroso. Las opiniones expresadas son interpretaciones del autor de la literatura científica publicada.